
Human Pangenome Project
The first human genome was generated from the international Human Genome Project in 2000, with major contributions by UC Santa Cruz. The current reference genome has been improved and annotated over the years, but is still an incomplete sequence and woefully inadequate as a representation of human diversity and genetic variation (as it is largely based mostly on one person’s genome).
Continue reading
The Human Pangenome Project will expand and diversify the human genome by using multiple sequencing technologies to generate more complete assemblies of 350 individuals that represent the greater genetic diversity of humanity. The project is funded by the National Human Genome Research Institute (NHGRI), part of the NIH. The project is driven by UC Santa Cruz, with major collaborators including the University of Washington in Seattle, Washington University in St. Louis, and Rockefeller University in New York.
The data and assemblies for this project will be completely open access and immediately available on NHGRI’s AnVIL cloud initiative that the CGP also is also developing. Members of the CGP will represent the Human Pangenome Project’s data coordination center and help to drive reproducible analysis methods and outreach for the project within the context of the AnVIL.
GENCODE
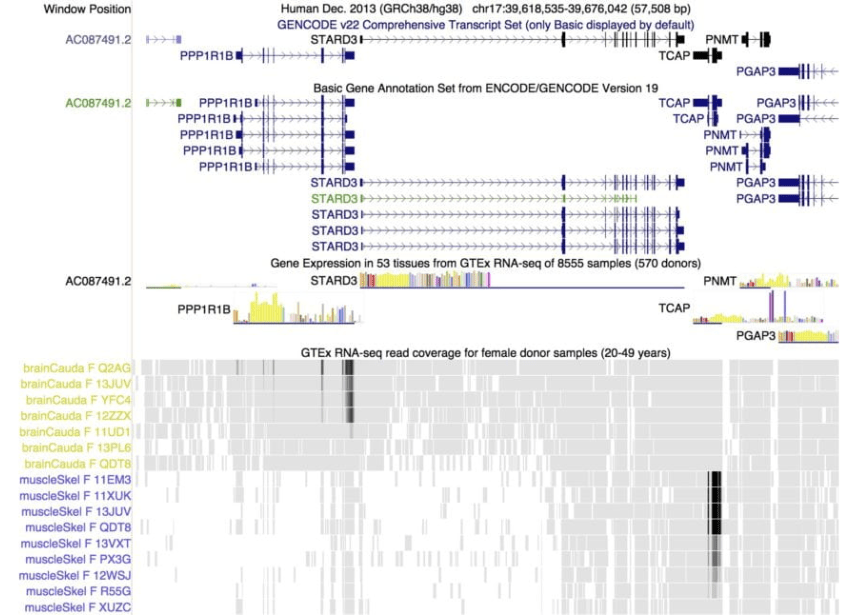
View of a 56 Kbp region of human chromosome 17 where GENCODE annotates one non-coding and 5 protein coding genes. Two genes in the region display tissue-specific gene expression as evidenced by GTEx RNA-seq. The TCAP (titin cap protein) is highly expressed in muscle tissue, while PP1R1B (a therapeutic target for neurologic disorders) shows expression in brain basal ganglia but not muscle.
The GENCODE project produces high quality reference gene annotation and experimental validation for human and mouse genomes.
Continue reading
The aims of the current GENCODE are:
- To continue to improve the coverage and accuracy of the GENCODE human gene set by enhancing and extending the annotation of all evidence-based gene features in the human genome at a high accuracy, including protein-coding loci with alternatively splices variants, non-coding loci and pseudogenes.
- To create a mouse GENCODE gene set that includes protein- coding regions with associated alternative splice variants, non-coding loci which have transcript evidence, and pseudogenes.
The mouse annotation data will allow comparative studies between human and mouse and likely improve annotation quality in both genomes. The process to create this annotation involves manual curation, different computational analysis and targeted experimental approaches. Putative loci can be verified by wet-lab experiments and computational predictions will be analysed manually.
The human GENCODE resource will continue to be available to the research community with quarterly releases of Ensembl genome browser (mouse data will be made available with every other release), while the UCSC genome browser will continue to present the current release of the GENCODE gene set.
The process to create this annotation involves manual curation, different computational analysis and targeted experimental approaches. Putative loci can be verified by wet-lab experiments and computational predictions will be analysed manually.
The Computational Genomics Lab, working with the wider consortium, is creating algorithms and software essential for finding and weighing the evidence for genes and isoforms.
(Adapted with permission from http://www.gencodegenes.org/)

GA4GH
The Global Alliance for Genomics and Health (GA4GH) is an international effort to promote, foster and standardize secure, ethical, privacy preserving sharing of genomic information for the betterment of global health outcomes. It has many, many organizations involved, all recognizing the importance of its mission, however it is largely a volunteer effort. To help ensure that it can meet its technical goals we are supporting with engineering effort a GA4GH integration group tasked with implementing and defining the necessary software standards.
Continue reading
Key to this is the creation of genomics Application Program Interfaces (APIs) to allow the exchange of genomic information across multiple organizations. The GA4GH standard is a freely available open standard for interoperability, which uses common web protocols to support the serving and sharing of data about nucleic acid sequences, variation, expression and other forms of genomic data. The API is implemented as a web service to create a data source that may be integrated downstream into visualization software, web-based genomics portals or processed as part of genomic analysis pipelines. Its goal is to overcome the barriers of incompatible infrastructure between organizations and institutions to enable DNA data providers and consumers to better share genomic data and work together on a global scale, advancing genome research and clinical application.

High Throughput Genomics Group
Genomics is in transition. The growth in data—driven by the need for vast sample sizes to gain statistical significance and the explosion of clinical sequencing—is far outpacing Moore’s law. Large projects like The Cancer Genome Atlas have generated petabyte scale datasets that very few groups have the capacity to analyze independently. Looking forward, improvements in the computing technologies will be insufficient to satisfy the community’s exponentially growing needs for computing throughput and storage capacity. The cost of computing on the rapidly growing data is compounded by the expanding complexity of genomic workflows. Typically, dozens of programs must be precisely configured and run to reproduce an analysis. The drastic increases in data volume and workflow complexity have created a serious threat to scientific reproducibility.
Continue reading
However, the shift to cloud platforms, the creation of distributed execution systems and the advent of lightweight virtualization technologies, such as containers, offers solutions to tackle these challenges. With UC Berkeley’s AMP lab we pioneered ADAM, a genomics platform built on Apache Spark that can radically improve the efficiency of standard genomic analyses. To support portable, scalable and reproducible workflows we have created Toil, a cross-cloud workflow engine that supports several burgeoning standards for workflow definition. We argue the flexibility afforded by such a system is not only efficient, but transformative, in that it allows the envisioning of larger, more comprehensive analyses, and for other groups to quickly reproduce results using precisely the original computations. To support scientific container discovery and sharing we are supporting Dockstore, a project pioneered by The Ontario Institute for Cancer Research (OICR), that is part of the Global Alliance for Genomics and Healtheffort that we are co-leading to develop APIs and standards for genomic containers.
Computational Nanopore Genomics
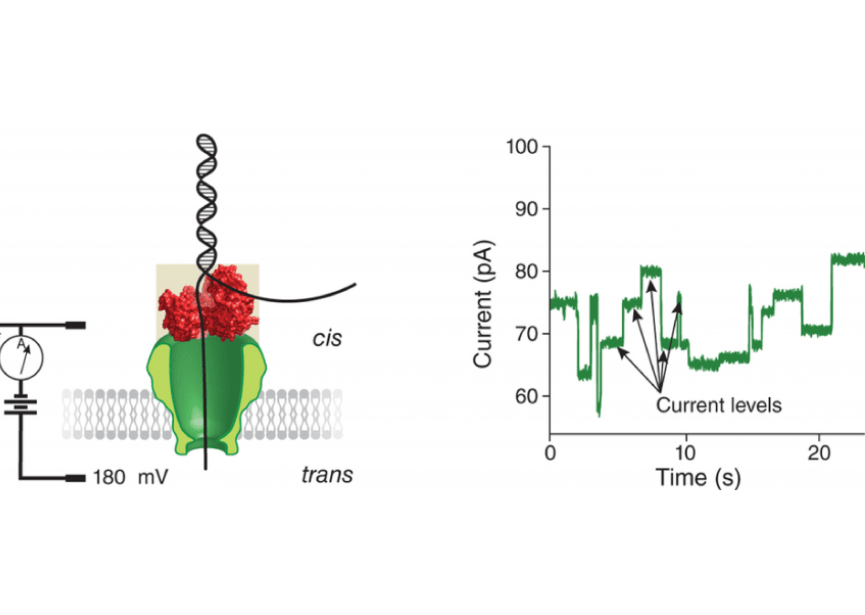
Nanopore technology is emerging as one of the leading contenders for next generation sequencing. The concept of nanopore sequencing was envisioned in the early 1990s by David Deamer (UC Santa Cruz) and Daniel Branton (Harvard). The recent commercialization of the technology, particularly the Oxford Nanopore’s disruptive MinION, has ignited the field. In collaboration with the UCSC Nanopore group, we are developing algorithms and code to analyze nanopore data. We foresee a time in the near future when low-cost nanopore sequencing can decode near complete genomes replete with methylation information and haplotype phasing.
Genome 10K
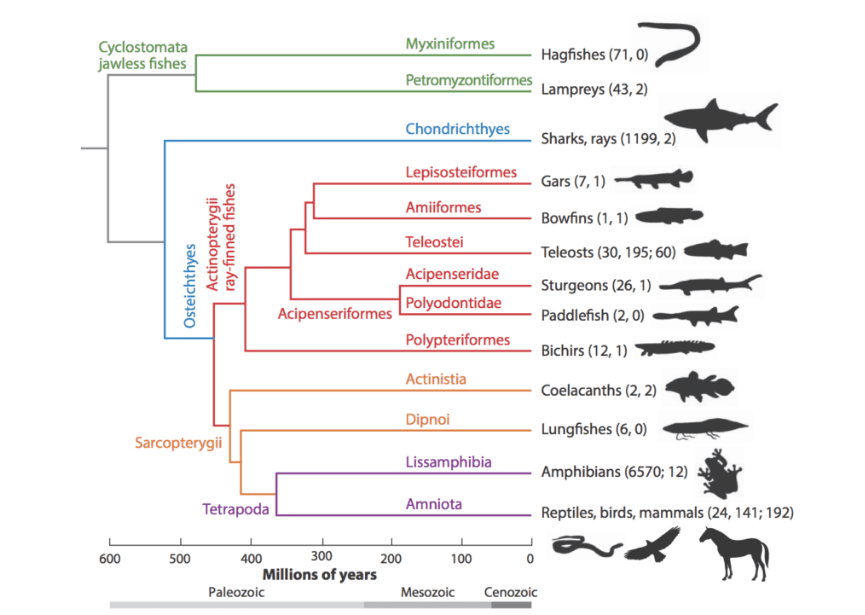
The vertebrate evolutionary tree
The Genome 10K project aims to assemble a genomic zoo—a collection of DNA sequences representing the genomes of 10,000 vertebrate species, approximately one for every vertebrate genus. The trajectory of cost reduction in DNA sequencing suggests that this project will be feasible within a few years. Capturing the genetic diversity of vertebrate species would create an unprecedented resource for the life sciences and for worldwide conservation efforts.
Continue reading
The growing Genome 10K Community of Scientists (G10KCOS), made up of leading scientists representing major zoos, museums, research centers, and universities around the world, is dedicated to coordinating efforts in tissue specimen collection that will lay the groundwork for a large-scale sequencing and analysis project.
The Computational Genomics Lab is invested in creating the algorithms and software to compare these vertebrates, allowing us to pinpoint the origin of the many diverse traits observed in our closest “siblings” in the history of life. The algorithms we are developing allow us to trace the history of evolution from the highest karyotype scale, dictating the structures of chromosomes, all the way down to tracing the ancestry of each individual base of DNA. This is an enormously complex and ambitious task, requiring an understanding of all the evolutionary processes that change genomes over time. It uses techniques from diverse areas of computer science and is deeply informed by the biology of genome evolution.
Tools and Platforms
The Computational Genomics Lab and Platform is responsible for developing and maintaining several important tools and platforms to help securely store, display, standardize, and analyze the enormous amounts of data involved in studies of human and non-human genomes. Our tools have provided the backbone for the Guinness World Record for sequencing speed, large scale comparisons of vertebrate genomes, the creation of the first ever draft of a human pangenome, and more.

Chan Zuckerberg Initiative (CZI) Human Cell Atlas (HCA) Data Sharing Platform
This ambitious, international project is attempting to create comprehensive reference maps of every single cell in the human body. The goal is to use this information to advance our understanding of diseases and how to treat them. The scale of the project’s ambition is matched only by the scope of its collaboration: Scientists from universities and institutes around the world, including UC Santa Cruz, the Wellcome Sanger Institute, the Broad Institute of MIT and Harvard, and the Chan Zuckerberg Initiative are joining forces, along with hundreds of participants and led by an organizing committee of 27 scientists from ten countries.
UCSC Xena
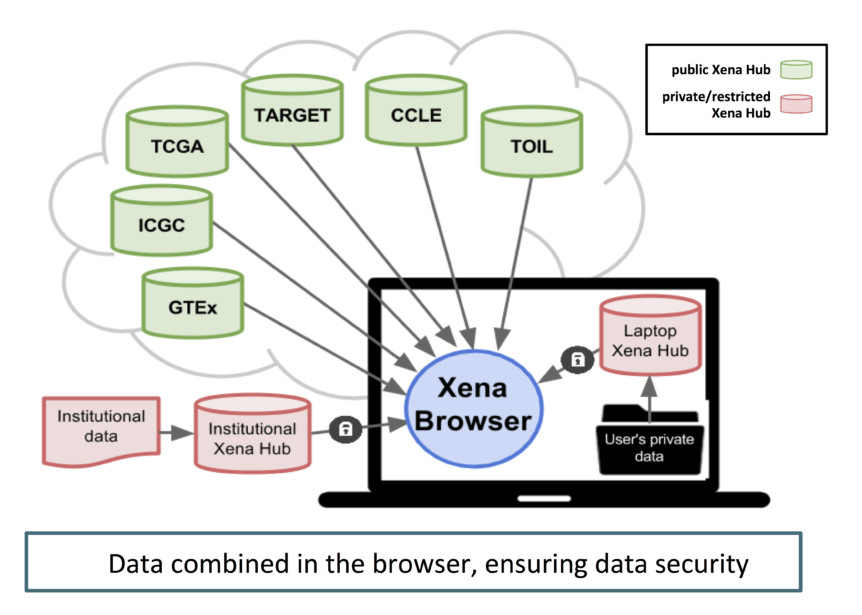
UCSC Xena is a bioinformatics tool to visualize functional genomics data from multiple sources simultaneously, including both public and private data. The Xena system consists of a set of federated data hubs and the Xena browser, which integrates across hubs, providing one location to analyze and visualize all data. The lightweight Xena data hubs are straightforward to install on Windows, Mac and Linux operating systems and easily allow hub administrators to authenticate users, ensuring that only authorized users have access to secure data. Loading data into a Xena hub is easy using either our application or command line interface. We host public data from major projects, such as TCGA, on public Xena hubs, giving users access without having to download these large datasets. While you can view data from each hub separately, the Xena system makes it easy to aggregate any arbitrary number of samples across many hubs, integrating public and/or private data into a ‘virtual cohort’. This allows researchers to combine new or preliminary results from their laptops or internal servers, or even data from a new paper, securely with vetted data from the public sphere.
Continue reading
Our public hubs host 806 public datasets from several large consortiums including TCGA (The Cancer Genome Atlas), ICGC (International Cancer Genome Consortium),Treehouse Childhood Cancer Project, CCLE (Cancer Cell Line Encyclopedia), and more. Xena hubs are flexible enough to handle most data types, including gene, exon, miRNA and protein expression, copy number, DNA methylation, somatic mutation, fusion and structural variant data along with phenotypes, subtype classifications and genomic biomarkers. All our data can be downloaded as slices or in large batches. Our browser instantaneously displays thousands of samples in many different visualization modes, including virtual spreadsheets, scatter plots and bar graphs. Dynamic Kaplan-Meier survival analysis helps investigators to assess survival stratification. Filter, sort, highlight and zoom to any samples of interest.

TOIL
Toil is a scalable, efficient, dynamic cross-platform pipeline management system written entirely in Python. Toil is being used by projects like the Treehouse childhood cancer initiative https://treehousegenomics.soe.ucsc.edu/ to create portable, scalable and reproducible analyses.
Toil was used to process over 20,000 RNA-seq samples in under four days using a commercial cloud cluster of 32,000 preemptable compute cores. Toil can also run workflows described in Common Workflow Language and supports a variety of schedulers, such as Mesos, GridEngine, LSF, Parasol and Slurm. Toil runs in the cloud on Amazon Web Services, Microsoft Azure and Google Cloud.See the Toil website and Toil Github for details.
Variation Graphs (vg)
- Watch our explainer videos.
- Visit our vg Github
Variation graphs are a pangenomic toolkit that provides a way to represent multiple genomes. Instead of displaying ten copies of ten completely different genomes, vg collapses the areas that are the same in all the sequences, something like 99%, and then displays bubbles in the graph to show where the sequences have variant base pairs. We’ve shown that we can use variation graphs to genotype difficult variants, and that this approach is efficient enough to scale to thousands of genomes. Variation graphs will be instrumental in creating a pangenome reference, a draft of which was released in May 2023.
Continue reading
A variation graph is composed of:
- nodes, which are labeled by sequences and ids
- edges, which connect two nodes via either of their respective ends
- paths, describe genomes, sequence alignments, and annotations (such as gene models and transcripts) as walks through nodes connected by edges
This model is similar to sequence graphs that have been used in assembly and multiple sequence alignment.
BRCA Exchange
We are developing the BRCA Exchange, a comprehensive, global data repository to catalogue variation within the BRCA genes and to collect individual-level evidence for their classification.
The scientific and medical communities have a tremendous and urgent need for a comprehensive data store of variants in the BRCA1 and 2 genes. Variation in these genes can indicate significant genetic predisposition to breast and ovarian cancer, leading causes of death that claim greater than fifty thousand lives in the United States annually. New evidence also implies mutations in BRCA1 and 2 are important in the development of other cancers, including prostrate and pancreatic cancer.
Continue reading
There is enormous public interest in BRCA testing, both because of growing public interest in genetic testing in general and following Angelina Jolie’s double mastectomy and hysterectomy and the ensuing “Angelina Effect.” Yet not all BRCA variants are pathogenic and most are of uncertain significance. There is therefore a scientific need to catalog all BRCA variants and evidence for their pathogenicity. Given the number of genetic testing companies and labs around the world that are contributing data to public repositories, there is also an unprecedented opportunity to build such a catalog.

BioData Catalyst (formerly DataSTAGE)
NHLBI BioData Catalyst is a cloud-based platform providing tools, applications, and workflows in secure workspaces. By increasing access to NHLBI datasets and innovative data analysis capabilities, BioData Catalyst accelerates efficient biomedical research that drives discovery and scientific advancement, leading to novel diagnostic tools, therapeutics, and prevention strategies for heart, lung, blood, and sleep disorders.
Though the primary goal of the BioData Catalyst project is to build a data science ecosystem, at its core, this is a people-centric endeavor. BioData Catalyst is also building a community of practice working collaboratively to solve technical and scientific challenges.

Dockstore
Dockstore, developed by the Cancer Genome Collaboratory, is an open platform for sharing Docker-based tools described with the Common Workflow Language used by the GA4GH.

AnVIL
The NHGRI Genomic Data Science Analysis, Visualization, and Informatics Lab-space (AnVIL) is a scalable and interoperable resource for the genomic scientific community, that leverages a cloud-based infrastructure for democratizing genomic data access, sharing and computing across large genomic, and genomic-related data sets.
Continue reading
The AnVIL facilitates integration and computing on and across large datasets generated by NHGRI programs, as well as initiatives funded by National Institutes of Health (NIH), or by other agencies that support human genomics research. In addition, the AnVIL is a component of the emerging federated data ecosystem and is expected to collaborate and integrate with other genomic data resources through the adoption of the FAIR (Findable, Accessible, Interoperable, Reusable) principles, as their specifications emerge from the scientific community. The AnVIL provides a collaborative environment, where datasets and analysis workflows can be shared within a consortium and be prepared for public release to the broad scientific community through AnVIL user interfaces. The AnVIL will be tailored for users that have limited computational expertise as well as sophisticated data scientist users.
Specifically, the AnVIL resource will provide genomic researchers with the following key elements:
- Cloud-based infrastructure and software platform
- Shared analysis and computing environment
- Interoperability and compliance with the emerging federated data ecosystem
- Cloud services cost control
- Genomic datasets, phenotypes and metadata
- Data access and data security
- User training and outreach
- Incorporation of scientific and technological advances for storing, accessing, sharing and computing on large genomic datasets